
![]()
|
|
|
|
MolDyn |
RUN program by the command
../$MDYN07HOME/mDynQ07 -i inProtcol -c inPDB [-mdR mdRestXYZVin] [-mv moveRes]
[-r1 inRestrainA1 ] [-r2 inRestrainA2] [-rB rigBodyFile]
[-sa saProtocol] [-mn molName] [-mdX mdFinalPDB] -o runOutFile [-er errorFile]
in parenthesis [ ] are uxilarry files. The auxilary files will be used by program if the main command file
defines respective task.
Command line DESCRIPTION:
-i inProtcol : file MdynPar.inp defines protocol for the mDyn particular Run
-c inPDB : file of the initial molecular structure as molec.pdb file in the PDB format
-mdR mdRestXYZVin : XYZ+Velocity file to REstart MD from the last snapshot file XYZV , see exaple t5
1arb.mdXYZVfin0001.pdb it is USED with $mdRestart keyword in command file
inProtcol
NOTE! the initial XYZ will be taken from mdRestXYZVin file !
the PDB file inPDB is not USED with the key -mdR
-r1 inRestrainA1 : file defines of positional restraints for atoms of the molecule
-r2 inRestrainA2 : file defines atom-atom distance restraints
-rB rigBodyFile : file defines rigid body segments of the main chain of protein
-mv moveRes : file defines List of moving Residues
-sa saProtocol : file defines simulated annealing protocol
-mn molName : character set defining molecula name. molName. will be attached to RESULT files
-o runOutFile : run output file
-mdX mdFinalPDB : final PDB file of the Energy/MD optimization
Current status of program run is printed on the standart output device (consol) or
can be redirected to user defined file or can be defined in the argument line:
-er errorFile : error message file : they are dublicated in the runOutFile
#
if file name definition in the argument line is missing for a file
than the default name is used for this file
NOTE! if the command line does not include a key -X , while the command file defines task which need data file coupled with -X keyword, than program try to find default (standart) name data file in the current directory.
Default names:
#
inProtcol = ./MdynPar.inp
inPDB = ./molec.pdb
mdRestXYZVfile = ./mdXYZVin.pdb
moveRes = ./moveRes.inp
inRestrainA1 = ./restrAt1.inp'
inRestrainA2 = ./restrAt2.inp'
rigBodyFile = ./rigBody.inp
saProtocol = ./SAprotocol.inp
molName = space
runOutFile = ./mDynSB.out
errorFile = ./mDynSB.err
mdFinalPDB = ./molMdFin.pdb
#
inProtcol = ./MdynPar.inp
The nain command file consist of lines with command keyword.
Keyword start with $ sign in the first position of line
One Keyword in line
#example of MdynPar.inp file and keyword description
# MdynPar.inp
$OUTfull ! full extended output of program run
#Initial PDB data quality
$Hread ! read INPUT pdb file with Hydrogens
! by default OUTshort option is ON
# DEfinition of OPtimized segments of protein:
$fullProtMD ! full molecule is flexible
#$MovingRes ! defines List of opimized segments
#FORCE FIELD MODIFICATIONS:
#
$shake=2 ! all valence bobds are fixed by shake method
$zeroRot ! exclude translation and rotation of the molecule
as rigid body
$hBond128 = 2.0 ! scaling coeff for H-bonds
! default=1.0 it is standart force field
$harmAt1PosRst=0.25 !invoke restraintsA1 type =
positional harmonic restraints for atom position
with harmConst (kcal/A^2).
program need a special file -r1 restrA1File
which defines restrained segmants of protein
see additional description
$distRestrA2 !invoke restraintsA2 type atom-atom distances
for user defined pairs of atoms in the file
-r2 restrA2File (see additional description)
$rigBody !invoke optimization with frozen internal structure of
protein main chain for user defined segments of sequence
need file -rB rigidBodySegment (see additional description)
$compactForce = 0.5 ! invoke additional compactization forces
! to accelerate protein folding
#
$aSoftCore = 0.5 !invoke SOFTNES for the van der waals atom-atom potential
! at the small (contact) atom-atom distances
! Use of the softCore VDW potential helps to optimize
! BAD molecular structures with many spartial atom-atom clashes
! values range 0 - 1 from very Soft to standart VDW
#SOLVATION MODEL
$SolvMod = GShell
#
#
# OPIMIZATION PROTOCOL:
$engCalc ! do energy calculation
$engOptim ! do energy optimization by local Optimizer
$nOptStep=1 !max N optim steps
#
#PROTOCOL for Molecular Dynamics:
$doMDyn ! do MolDynamics
$MDSA !do MolecularDynamis SimAnnealing
needs SAprotocolFile -sa saProtocol File,
see additional description
#
#PROTOCOL of MD equilibration:
#
$initMDTemp=50.00 !initial Temperature to start MolDyn
$bathMDTemp=50.00 !thermostat temperature of thermostat i.e. target temperature
$runMDnstep=2000 !number of time-steps for MD simulation
$mdTimeStep=0.002
#
$NTV=1 ! MD ensemble definition
#
#
# MD Trajectory writing:
$nwtra=500
$WRpdb ! write snarshort structures in the PDB format
! default WRpdbq OPTion is ON : extended PDB format
! PDB + Qatom
#
END
#
NOTE that parameter file formatted, i.e. $ sign should be the firs character of the line
----------------------------------------------------------------------------
KEYWORD LIST:
keyw = 'OUTfull'
keyw = 'WRpdb'
keyw = 'Hread'
keyw = 'fullProtMD'
keyw = 'MovingRes'
keyw = 'LigRes'
keyw = 'doLigDock'
keyw = 'MDSA'
keyw = 'SolvMod'
keyw = 'zeroRot'
keyw = 'hBond128'
keyw = 'harmAt1PosRst'
keyw = 'distRestrA2'
keyw = 'compactForce'
keyw = 'shake'
keyw = 'engCalc'
keyw = 'engOptim'
keyw = 'nOptStep'
keyw = 'aSoftCore'
keyw = 'initMDTemp'
keyw = 'bathMDTemp'
keyw = 'mdTimeStep'
keyw = 'runMDnstep'
keyw = 'doMDyn'
keyw = 'mdRestart'
keyw = 'NTV'
keyw = 'nwtra'
-----------------------------------------------------------------------------
KEYWORD DESCRIPTION:
#OUTPUT DETAILES:
$OUTfull ! full extended output of program run
! by default OUTshort option is ON
#
# INPUT PDB FILE DETAILES:
$Hread ! defines that all Hydrogens will be read from input molecule structure -c inPDB file
otherwise the ALL HYDrogens will be restored by the program, i.e.
all H atoms will be deleted and added according to molecular topology for RESidues.
Using Library in the ./dat/h_add.dat
NOTE! it is recommended start to works with a new protein without option $Hread even if the PDB
file has all hydrogen atoms, because the hydrogen atom names for protein side chains
have multiple definition in the PDB data base.
It is better if mDyn program will add all hydrogens to the heavy atoms.
#DEFINITION OF OPTIMIZED RESIDUES:
$fullProtMD !defines FULL (i.e. ALL atoms) of the USER molecule
will be free to move in energy relaxation or molDyn
$MovingRes ! logical keyWord defines that only a defined set of RESidue are free
this keyWord is coupled with file -mv moveRes in the argument line to start
the program
default name for moveRes file is ./moveRes.inp
#EXAMPLE of ./moveRes.inp
#1arb
aaaaaaIIIIiiii
#
MOVRES 1 10 !line defines first and last resudue of moving segments integers devided by space
MOVRES 45 76
MOVRES 115 260
end !end or END should be last line if the file
************
#FORCE FIELD DEFINITION:
$hBond128 = 2.0 ! scaling coeff for H-bonds
$aSoftCore = 0.5 !invoke van der waals atom-atom potential with modified
! SoftCore at the small (contact) atom-atom distances
! SoftCore modification is used for energyOPtimization
and MD equilibration stages.
! Use of the softCore VDW potential helps to optimize
! BAD structures with many starical atom-atom clashes
! values range 0 - 1 from very Soft to standart VDW
$harmAt1PosRst=0.25 ! digital keyWord define RESidue segments with 1 atom position harmonic
restrants.
0.25 = harmonic restrain Constant K
restrEnergy = 0.5*K(r - r0)**2,
the reference position r0 = initialXYZinput.pdb - positions from
the initial INPut PDB file which defines INItial structure of molecule
this keyWord is coupled with file -r1 inRestrainA1 of the argument line to start
the program mdyn
default name for inRestrain file is ./restrAt1.inp
#EXAMPLE of inRestrainA1 file:
#harmonically restrained RESidue segments
#xxxxxIIIIiiiiaaAAA
#(6x,2i4,a40)
RESTA1 1 63 PBB ! line starts from keyWord RESTAT numbers=first/last residue of segment
! PBB (only protein backbone atoms are restrained, i.e. side chains are free)
RESTA1 78 120 ALL ! ALL (all atoms are restrained)
! integers and words are devided by space
end
# ---------------------------------------------------
$distRestrA2 ! defines optimization/MD with atom-atom dist RestrainA2
! needs file [-r2 inRestrainA2] in command line
-r2 inRestrainA2 : default name : restrAt2.inp
#
EXAMPLE of inRestrainA2 file:
#harmonically restrained Atom-Atom distances
#xxxxxx
#keyword atom1 atom2 distA HarmConst(kcal/mol*A^2)
RESTA2 ND2 ASN 222 : OG1 THR 219 = 7.0 1.5
RESTA2 O GLY 170 : OG1 THR 219 = 8.0 2.5
RESTA2 OH TYR 109 : OG1 THR 111 = 7.5 3.0
END
#----------------------------------------------------
$rigBody !defines optimizatiom/MD considering some segments of the main chain
! as a rigid body.
! The List of rigid segments of the main chain is user defined.
! Each segment will keep rigid internal structure of the protein main chain,
! has rotatational and translational degrees of freedom.
! The side chains of the rigid segments are flexible.
#Needs file rigidBody.inp
#EXAMPLE of rigidBody.inp file:
#
RIGB01 11 16 !line defines first and last resudue of moving segments integers devided by space
RIGB02 47 59
RIGB03 77 99
end !end or END should be last line if the file
# - - - - - - - - - - - - - - - - - - - - - - - - -
$compactForce = 0.25 ! define additional compactization forces for protein atoms
! Recomended forceParameter = 0.1 - 1.0
# --------------------------------------------------
$shake=2 ! invoke shake subroutine to keep bonds fixed. =1 -bonds with Hydrogen, =2 all bonds
----------------------------------------------------
#Defining of the SOLVation model:
there are 4 variants of Implicit models
1 variant of Explicit model
#:
$SolvMod = GShell ! implicit Gaussian Shell solvation model
$SolvMod = GShell + WBrg ! implicit Gaussian Shell solvation model + WaterBridges between polar atoms
! WaterBridges descride solvent mediated interactions trough stong bound water
! molecules via implicit model of water bridges
$SolvMod = GBorn ! implicit Generalized Born model + SAS HydroPhobic solvation
$SolvMod = GBorn + WBrg ! implicit Generalized Born model + SAS HydroPhobic solvation + WaterBridges
$Solv = ExWshell 4.5 [A] ! explicit water shell of 4.5 Angst around protein;
! recomended thikness 3.0 - 6.0 A
---------------------------------------------------
$mdRestart ! restart molDynamics from a snapshot [molName.]mdXYZVfin000N.pdb
the file [molName.]mdXYZVfin000N.pdb should be copied to the file mdyn Restart file
mdXYZVin.pdb
$doMDyn ! do molecular dynamics
$MDSA ! do Molecular Dynamical Simulated Annealing
! coupled with file -sa SAprotocol which define protocol of the simulated annealing
#EXAMPLE of Aprotocol.inp file
#SA protocol
#nSAstep 2
#(f10.1,1x,f8.1,1x,3(f6.1,1x)
# nMDstep tempTg SCvdW wfHb128BB wfhB128BS
SAPROT 100000 500.0 0.8 1.0 1.0 !line starts from keyword SAPROT
SAPROT 100000 100.0 1.0 1.0 1.0
END
#
nMDstep - number of md timeStep
tempTg - target temperature in K, this temperature will be reach during ntimeMX steps
SCvdW - parameter 0 - 1 to define softness of the van der waals potential. Soft potential
modifies Potential Energy Surface and decrease barriers of conformational transitions
wfHb128BB,
wfhB128BS - (1 - 0) scaling factors for BackBone-BackBone and
BackBone-SideChain Hydrogen Bond energy
#--------------------------------------------------
#
# OPIMIZATION PROTOCOL:
$engCalc ! do energy calculation
$engOptim ! do energy optimization by local Optimizer
$nOptStep=1 !max N optim steps
#
#PROTOCOL for Molecular Dynamics:
$doMDyn ! do MolDynamics
$MDSA !do MolecularDynamis SimAnnealing
needs SAprotocolFile -sa saProtocol File,
#MD EQUILIBRATION:
$initMDTemp=50.00 !defines initial temperature to start MD
! recommended low temperature < 50K
! temperature can be steadelly increased to the 300K and higher
! USING $MDSA option
$bathMDTemp=50.00 ! bath temperature in the MD equilibration run
$runMDnstep=2000 ! number of MD time steps in the equilibration run
$mdTimeStep=0.002 ! value of the MD time step in ps,
! recomended 0.001 - 0.002
$NTV=1 ! ansemble NTV=0/1
! =1 md run with constant T
#MD TRAJECTORY WRITING
$nwtra=500 ! structure XYZ (snapshot) will be written
!as a series of molMdResXXXX.pdb files
$WRpdb ! write snapshort structures in the PDB format
! default is WRpdbq OPTion is ON : extended PDB format
! PDB + Qatom column
#* * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
#
-c inPDB file - standart pdb file
#EXAMPLE of inPDB file:
****************************************************************************************
NOTE! it is recommended to start to work with a new protein without option $Hread even if the PDB
file has all hydrogen atoms, because the hydrogen atom names for protein side chains
have multiple definition in the PDB data. It is better if mDyn program will add all hydrogens
to the heavy atoms.
*******************************************************************************************
REMARK: PDB:
ATOM 1 N GLY A 1 11.726 -10.369 10.598
ATOM 2 H1 GLY A 1 11.921 -11.015 9.807
ATOM 3 H2 GLY A 1 12.518 -10.395 11.271
ATOM 4 H3 GLY A 1 10.852 -10.663 11.079
ATOM 5 CA GLY A 1 11.567 -9.015 10.090
ATOM 6 HA2 GLY A 1 10.772 -8.977 9.420
ATOM 7 HA3 GLY A 1 12.439 -8.710 9.612
ATOM 8 C GLY A 1 11.280 -8.099 11.303
ATOM 9 O GLY A 1 11.256 -8.584 12.493
ATOM 10 N VAL A 2 11.060 -6.876 11.020
ATOM 11 H VAL A 2 11.066 -6.574 10.025
etc.
TER ! CHAIN TERmination
ATOM 1302 N GLY A 94 10.957 -15.678 12.832
ATOM 1303 H GLY A 94 10.735 -14.663 12.877
ATOM 1303 H GLY A 94 10.735 -14.663 12.877
ATOM 1304 CA GLY A 94 10.193 -16.559 11.950
ATOM 1305 HA2 GLY A 94 9.428 -16.004 11.516
ATOM 1306 HA3 GLY A 94 9.784 -17.323 12.525
ATOM 1307 C GLY A 94 11.016 -17.184 10.843
...
etc.
TER ! CHAIN TERmination
END ! file END
* * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
#
# PDB mDyn trajectory file description:
#
Program mDyn generate a series of snapshot files, e.g., 1arb.molMdRes0nnn.pdb (test/t4)
the molMdResXXXX.pdb file (see example) contains all atomic coordinates and additional information
in the REMARK: lines
####
REMARK: Md result : MdTime(ps): 2.4940
REMARK: $nstep: 1247
REMARK: $nRecPDB: 5
REMARK: RMSD(x0): 0.43 <- RMSD all atom
REMARK: badBond: n,erAv(A) : 0 0.000 <- number and error Average for bond length in Angstrem
REMARK: badAng : n,erAv(grd): 8 9.42 <- number and error Average for bond angles in grad
# ENERGY TERMS for the given structure
REMARK: $ENERGY: :Kcal
REMARK: eVbondDef: 100.89315 <-bond deformation energy
REMARK: eVangDef : 441.63705 <-angle deformation energy
REMARK: eImpDef : 35.68147 <-Improper torsion agle [planarity] energy
REMARK: eTorsDef : 691.25769 <-torsion potentioal energy
REMARK: engVDWR1 : -1031.16211 <- van der waals energy for cutoff R1=8 A
REMARK: ehBHxY128: -608.70599 <- H-bondinds energy
REMARK: engCOULR1: -816.25323 <- COULOMBIC for distances < cutoff R1
REMARK: engCOULR2: -4.47208 <- COULOMBIC for distances Rij, R1< rij
To run Ligand docking modules, the main command file MdynPar.inp have to include the next keywords: # keywords=value $LigRes= 282 283 !define start/end ligandResidues
in the inPDB file [(i4,1x,i4) format after= ] !the residues numbers are the same as it is in the initial !inPDB file [united pdb file of protein + ligand] $doLigDock=1 !run docking for USER defined initial position of Ligand ! as it is in the initial inPDB file [united pdb file of protein + ligand] ! Docking is done via simulated annealing molDynamics ! with coupled temperature and force field variation. ! Ligand CMass can move in vicinity of initial ! position +/- 4.0 A ! Orientational global optimization are done via ! simulated annealing MD with multiple start ! orientations. Initial orientations are uniformly ! cover all orientational phase space with distance = 90 deg $doLigDock=2 ! run ab initio docking ! This option will seach all binding sites on the ! protein molecular surface including cavities and crevices. ! 1) search of surface cavities, crevicies and groovs ! 2) calculation and scoring of binding site candidate ! positions based on the number of ligand-protein atom-atom contacts. ! 3) ligand docking by simulated annealing molecular dynamics for best ! candidate binding sites. #REMARKS: 1) -c inPDBfile in command line should include proteinXYZ + ligandXYZ. it is recomended to make initial Ligand XYZ in the file inPDBfile in a contact vicinity of Protein. 2) For a new Ligand, the Ligand molecular topology SHOULD BE included into the LIBrary topology file bs_one_all94.dat at the moment the topology LIB includes the next Ligands # DOCK TEST#1 ./1bty - trypsine-benzamidine complex 1bty.ben.Native.pdb - protein-lig complex with NATIVE binding mode for Ligand 1bty.ben.notNative.pdb - protein-lig complex with notNATIVE (arbitryry) mode for Ligand the both files can be used as inPDB file MdynPar_1bty.inp moveRes_1bty.inp SAprotocol_1bty.inp : INPUT files to define JOB protocol # #Run the mDyn program: > $MDYN07HOME/mDynQ07 -i 1bty_MdynPar.inp -c 1bty.ben.notNative.pdb -sa SAprotocol_1bty.inp -mv moveRes_1bty.inp -mn 1bty -o 1bty.out # UNDESTANDING DOCKING RESULTS: Program creates a files in current job directory: 1) file 1bty.bSiteAtOnSAS00.pdb shows XYZ positions of binding site candidates on the protein surface scored by contact score ~ number of protein atoms in a vicinyty of the binbing site. #LigBindGridOnSAS: bindSite XYZ contactScore ATOM 1 LBSt 1 16.536 26.130 8.764 11 ATOM 2 LBSt 2 29.319 14.972 16.378 11 ATOM 3 LBSt 3 6.595 15.454 32.366 9 ATOM 4 LBSt 4 28.049 26.396 3.572 9 ATOM 5 LBSt 5 37.370 14.662 29.278 8 ATOM 6 LBSt 6 9.605 28.662 39.481 7 ATOM 7 LBSt 7 18.280 35.574 15.402 7 ATOM 8 LBSt 8 30.648 34.679 44.060 7 ATOM 9 LBSt 9 34.040 33.767 21.484 7 ATOM 10 LBSt 10 5.056 19.922 18.987 6 ATOM 11 LBSt 11 25.308 5.865 13.437 6 ATOM 12 LBSt 12 13.241 31.812 30.019 6 ... ATOM 40 LBSt 40 25.260 6.929 29.909 4 ATOM 41 LBSt 41 26.781 13.047 43.008 4 Docking alhorithm put ligand center into each of the positions with SCore >= 6 and refine ligand position&orientation and conformation via global optimization by Simulated annealing coupled with protein-Ligand force field deformation. # The resulting ligand positions are collected in the files : 1bty.LigDockFin000.001.pdb 1bty.LigDockFin000.002.pdb 1bty.LigDockFin000.003.pdb 1bty.LigDockFin001.001.pdb 1bty.LigDockFin001.002.pdb 1bty.LigDockFin001.003.pdb ... 1bty.LigDockFin015.003.pdb # File 1bty.LigDockFin_ePL.res : collects the refined total Potential energy of Lig-Lig + Lig-Prot interactions for Ligand Docking modes: # NN LigDockFinXXX.XXX.pdb ePLtot eVDW eCoul eHb eGeo eSolv tempTAv 1 ./LigDockFin000.001.pdb -26.2 -4.9 -7.7 -9.6 -6.1 2.2 56.1 2 ./LigDockFin000.002.pdb -25.7 -2.9 -9.6 -9.7 -6.3 2.8 56.1 3 ./LigDockFin000.003.pdb -25.3 -3.7 -8.8 -9.6 -5.8 2.7 56.1 4 ./LigDockFin001.001.pdb -25.9 -9.0 -1.7 -9.5 -10.2 4.5 56.9 5 ./LigDockFin001.002.pdb -21.6 -9.3 0.7 -4.6 -11.5 3.2 56.9 6 ./LigDockFin001.003.pdb -21.5 -9.7 -0.5 -9.1 -4.3 2.2 56.9 7 ./LigDockFin002.001.pdb -42.5 -18.6 -8.1 -11.2 -8.6 3.9 51.9 !best1 mode =native 8 ./LigDockFin002.002.pdb -42.4 -21.0 -6.6 -10.5 -8.0 3.8 51.9 9 ./LigDockFin002.003.pdb -42.0 -18.5 -8.6 -11.9 -8.2 5.2 51.9 10 ./LigDockFin003.001.pdb -15.7 -6.7 1.2 -4.9 -7.3 2.0 46.2 11 ./LigDockFin003.002.pdb -14.9 -7.5 1.2 -4.8 -7.2 3.5 46.2 12 ./LigDockFin003.003.pdb -13.7 -5.7 2.1 -4.4 -8.2 2.6 46.2 13 ./LigDockFin004.001.pdb -26.1 -4.7 -8.1 -9.8 -6.2 2.6 49.8 14 ./LigDockFin004.002.pdb -25.5 -6.1 -6.1 -9.6 -6.0 2.3 49.8 15 ./LigDockFin004.003.pdb -25.5 -4.8 -7.0 -9.8 -5.2 1.3 49.8 16 ./LigDockFin005.001.pdb -31.3 -12.0 -5.0 -9.5 -7.7 2.9 55.5 17 ./LigDockFin005.002.pdb -30.7 -11.2 -5.0 -9.7 -7.4 2.6 55.5 18 ./LigDockFin005.003.pdb -30.7 -12.3 -5.0 -9.5 -7.3 3.4 55.5 19 ./LigDockFin006.001.pdb -19.8 -7.7 -2.7 -4.9 -7.1 2.6 49.3 20 ./LigDockFin006.002.pdb -18.7 -10.2 -2.5 -4.8 -7.9 6.6 49.3 21 ./LigDockFin006.003.pdb -18.7 -9.1 1.8 -7.9 -7.4 3.8 49.3 22 ./LigDockFin007.001.pdb -20.9 -8.8 0.5 -9.6 -5.5 2.5 58.5 23 ./LigDockFin007.002.pdb -20.7 -8.9 1.2 -10.5 -5.6 3.2 58.5 24 ./LigDockFin007.003.pdb -20.1 -7.6 1.3 -10.8 -4.9 2.0 58.5 the file 1bty.LigDockFin*.pdb also keeps the final potential energies of docking modes: # 1bty.LigDockFin000.001.pdb:engPOTENTLG: -24.76501 1bty.LigDockFin000.002.pdb:engPOTENTLG: -24.53164 1bty.LigDockFin000.003.pdb:engPOTENTLG: -24.44349 1bty.LigDockFin001.001.pdb:engPOTENTLG: -24.03668 1bty.LigDockFin001.002.pdb:engPOTENTLG: -22.61240 1bty.LigDockFin001.003.pdb:engPOTENTLG: -20.96410 1bty.LigDockFin002.001.pdb:engPOTENTLG: -40.25522 !best docking mode 1bty.LigDockFin002.002.pdb:engPOTENTLG: -40.24239 1bty.LigDockFin002.003.pdb:engPOTENTLG: -40.21294 1bty.LigDockFin003.001.pdb:engPOTENTLG: -13.18473 1bty.LigDockFin003.002.pdb:engPOTENTLG: -12.79959 1bty.LigDockFin003.003.pdb:engPOTENTLG: -12.70576 1bty.LigDockFin004.001.pdb:engPOTENTLG: -24.15409 1bty.LigDockFin004.002.pdb:engPOTENTLG: -23.90785 1bty.LigDockFin004.003.pdb:engPOTENTLG: -23.87370 1bty.LigDockFin005.001.pdb:engPOTENTLG: -28.31307 1bty.LigDockFin005.002.pdb:engPOTENTLG: -28.17037 1bty.LigDockFin005.003.pdb:engPOTENTLG: -28.14446 1bty.LigDockFin006.001.pdb:engPOTENTLG: -20.06244 1bty.LigDockFin006.002.pdb:engPOTENTLG: -18.94587 1bty.LigDockFin006.003.pdb:engPOTENTLG: -18.48825 1bty.LigDockFin007.001.pdb:engPOTENTLG: -18.42872 1bty.LigDockFin007.002.pdb:engPOTENTLG: -18.37239 ..... 1bty.LigDockFin013.001.pdb:engPOTENTLG: -21.23134 1bty.LigDockFin013.002.pdb:engPOTENTLG: -20.48702 1bty.LigDockFin013.003.pdb:engPOTENTLG: -19.79687 1bty.LigDockFin014.001.pdb:engPOTENTLG: -17.41916 1bty.LigDockFin014.002.pdb:engPOTENTLG: -17.05084 1bty.LigDockFin014.003.pdb:engPOTENTLG: -16.60287 1bty.LigDockFin015.001.pdb:engPOTENTLG: -21.14995 1bty.LigDockFin015.002.pdb:engPOTENTLG: -20.88982 1bty.LigDockFin015.003.pdb:engPOTENTLG: -20.61083 # **************************************************************** # # DOCK TEST#2 alpha-thrombin/bemzamidine complex : 1dwb # 1dwb.ben.Native.pdb 1dwb.ben.notNative.pdb - two variants of inPDB file # MdynPar_1dwb.inp moveRes_1dwb.inp SAprotocol_1dwb.inp : input Job Protocol files # file 1dwb.LigDockFin.ePL.res: - total potential energy of Lig-Lig + Lig-Prot interactions: # 1dwb.LigDockFin000.001.pdb:engPOTENTLG: -19.92194 1dwb.LigDockFin000.002.pdb:engPOTENTLG: -19.81434 1dwb.LigDockFin000.003.pdb:engPOTENTLG: -19.30406 1dwb.LigDockFin001.001.pdb:engPOTENTLG: -43.80969 !best docking mode 1dwb.LigDockFin001.002.pdb:engPOTENTLG: -43.79070 1dwb.LigDockFin001.003.pdb:engPOTENTLG: -43.69997 1dwb.LigDockFin002.001.pdb:engPOTENTLG: -21.91559 1dwb.LigDockFin002.002.pdb:engPOTENTLG: -18.90804 1dwb.LigDockFin002.003.pdb:engPOTENTLG: -18.59642 1dwb.LigDockFin003.001.pdb:engPOTENTLG: -30.16282 1dwb.LigDockFin003.002.pdb:engPOTENTLG: -25.75561 1dwb.LigDockFin003.003.pdb:engPOTENTLG: -25.32805 .. *************************************************************************************** #DOCK TEST#3 ./1dwc - alpha-Thrombin/MIT ligand complex # inPDB file: Job protocol files: MdynPar_1dwc_d2.inp moveRes_1dwc.inp SAprotocol_1dwc_03.inp # Docking result potential energy file: 1dwc.ePL.LigDockFin.res: # LigDockFin000.001.pdb:engPOTENTLG: -53.51793 !refined Native Binding mode LigDockFin000.002.pdb:engPOTENTLG: -52.98364 LigDockFin000.003.pdb:engPOTENTLG: -45.18925 LigDockFin001.001.pdb:engPOTENTLG: -50.56367 !best2 docking result = native mode (differenr Lig conformation) LigDockFin001.002.pdb:engPOTENTLG: -49.67875 LigDockFin001.003.pdb:engPOTENTLG: -47.75756 LigDockFin002.001.pdb:engPOTENTLG: -29.34865 LigDockFin002.002.pdb:engPOTENTLG: -28.29562 LigDockFin002.003.pdb:engPOTENTLG: -27.49329 LigDockFin003.001.pdb:engPOTENTLG: 158.53473 LigDockFin003.002.pdb:engPOTENTLG: 180.83168 LigDockFin003.003.pdb:engPOTENTLG: 214.47627 LigDockFin004.001.pdb:engPOTENTLG: -35.89280 LigDockFin004.002.pdb:engPOTENTLG: -33.39799 LigDockFin004.003.pdb:engPOTENTLG: -32.96119 ... LigDockFin029.001.pdb:engPOTENTLG: -25.19725 LigDockFin029.002.pdb:engPOTENTLG: -21.06659 LigDockFin029.003.pdb:engPOTENTLG: -18.58114 LigDockFin030.001.pdb:engPOTENTLG: -52.92000 !best1 docking mode = native mode LigDockFin030.002.pdb:engPOTENTLG: -45.43426 LigDockFin030.003.pdb:engPOTENTLG: -41.80339 ... **************************************************************************************** # DOCK TEST#4 ./1stp : complex streptavidine/biotin # inPDB files: 1stp.btn.notNative.pdb 1stp.btn.Native.pdb Job protocol files: MdynPar_1stp.inp moveRes_1stp.inp SAprotocol_1stp.inp # Docking result potential energy file: 1stp.LigDockFin.ePL.res # 1stp.LigDockFin000.001.pdb:engPOTENTLG: -20.67740 1stp.LigDockFin000.002.pdb:engPOTENTLG: -18.70130 1stp.LigDockFin000.003.pdb:engPOTENTLG: -16.49737 1stp.LigDockFin001.001.pdb:engPOTENTLG: -59.33578 !best docking MODE = native 1stp.LigDockFin001.002.pdb:engPOTENTLG: -58.89420 1stp.LigDockFin001.003.pdb:engPOTENTLG: -58.77360 1stp.LigDockFin002.001.pdb:engPOTENTLG: -24.83950 1stp.LigDockFin002.002.pdb:engPOTENTLG: -20.99019 .. **************************************************************************************** # #DOCK TEST#5 ./3tpi : complex Trypsinogen/ILE-VAL dipeptide # inPDB file : 3tpi.IV.notNative.pdb Job protocol files: MdynPar_3tpi.inp moveRes_3tpi.inp SAprotocol_3tpi.inp # Docking result potential energy file: 1stp.LigDockFin.ePL.res: # LigDockFin000.001.pdb:engPOTENTLG: -75.41046 LigDockFin000.002.pdb:engPOTENTLG: -74.73651 LigDockFin000.003.pdb:engPOTENTLG: -73.62763 LigDockFin001.001.pdb:engPOTENTLG: -76.07257 !best docking mode = native LigDockFin001.002.pdb:engPOTENTLG: -75.64239 LigDockFin001.003.pdb:engPOTENTLG: -75.56549 LigDockFin002.001.pdb:engPOTENTLG: -39.38153 LigDockFin002.002.pdb:engPOTENTLG: -37.80946 LigDockFin002.003.pdb:engPOTENTLG: -37.19353 LigDockFin003.001.pdb:engPOTENTLG: -44.83166 LigDockFin003.002.pdb:engPOTENTLG: -44.66529 LigDockFin003.003.pdb:engPOTENTLG: -44.60099 LigDockFin004.001.pdb:engPOTENTLG: -74.91846 !best2 docking mode ~ native: VALine side chain change conformation LigDockFin004.002.pdb:engPOTENTLG: -74.83774 LigDockFin004.003.pdb:engPOTENTLG: -73.65620 LigDockFin005.001.pdb:engPOTENTLG: -35.99043 LigDockFin005.002.pdb:engPOTENTLG: -34.74246 LigDockFin005.003.pdb:engPOTENTLG: -34.45740 LigDockFin006.001.pdb:engPOTENTLG: -34.24403 LigDockFin006.002.pdb:engPOTENTLG: -33.57267 LigDockFin006.003.pdb:engPOTENTLG: -33.01848 LigDockFin007.001.pdb:engPOTENTLG: -36.43544 ... # **************************************************************************************** #DOCK TEST#6 ./1hvr - complex HIV-1 protease/XK263 ligand : 1hvr PdB code # inPDB file : 1hvr.NativeMode.pdb job protocol files: MdynPar_1hvr.inp moveRes_1hvr.inp SAprotocol_1hvr.in # Docking result potential energy file: 1hvr.ePL.LigDockFin.res: LigDockFin000.001.pdb:engPOTENTLG: -72.49685 ! md optimized native binding mode LigDockFin000.002.pdb:engPOTENTLG: -67.88530 LigDockFin000.003.pdb:engPOTENTLG: -67.32499 LigDockFin001.001.pdb:engPOTENTLG: -75.05936 !** best1 docking mode = native LigDockFin001.002.pdb:engPOTENTLG: -74.41620 LigDockFin001.003.pdb:engPOTENTLG: -72.38953 LigDockFin002.001.pdb:engPOTENTLG: -27.65386 LigDockFin002.002.pdb:engPOTENTLG: -24.26098 LigDockFin002.003.pdb:engPOTENTLG: -23.37482 LigDockFin003.001.pdb:engPOTENTLG: -40.34463 LigDockFin003.002.pdb:engPOTENTLG: -35.47597 LigDockFin003.003.pdb:engPOTENTLG: -34.07937 ... LigDockFin008.001.pdb:engPOTENTLG: -15.30777 LigDockFin008.002.pdb:engPOTENTLG: -10.51002 LigDockFin008.003.pdb:engPOTENTLG: -3.76347 LigDockFin009.001.pdb:engPOTENTLG: -64.55802 !best2 docking mode = native like LigDockFin009.002.pdb:engPOTENTLG: -64.05453 LigDockFin009.003.pdb:engPOTENTLG: -36.51418 LigDockFin010.001.pdb:engPOTENTLG: -18.38447 ... # ************************************************************************************* # #DOCK TEST#7 ./4phv Complex HIV-1 protease with inhibitor VAC : PDB code 4phv # inPDB file: 4phv.NativeMode.pdb job protocol files: MdynPar_4phv.inp moveRes_4phv.inp SAprotocol_4phv.inp # Docking result potential energy file: 4phv.ePL.LigDockFin.res: # LigDockFin000.001.pdb:engPOTENTLG: -103.07942 !optimized NAtive binding mode LigDockFin000.002.pdb:engPOTENTLG: -102.91592 LigDockFin000.003.pdb:engPOTENTLG: -87.68965 LigDockFin001.001.pdb:engPOTENTLG: -96.85534 !best1 docking mode ~= native LigDockFin001.002.pdb:engPOTENTLG: -86.03489 LigDockFin001.003.pdb:engPOTENTLG: -72.52062 LigDockFin002.001.pdb:engPOTENTLG: -73.35270 !best2 docking mode : perturbed Lig conf. LigDockFin002.002.pdb:engPOTENTLG: -73.13903 LigDockFin002.003.pdb:engPOTENTLG: -72.76502 LigDockFin003.001.pdb:engPOTENTLG: -57.68087 LigDockFin003.002.pdb:engPOTENTLG: -57.64718 LigDockFin003.003.pdb:engPOTENTLG: -57.60190 ... # ********************************************************************************************** #DOCK TEST#8 ./1hiv Complex HIV-1 protease with inhibitor NOA Ligand (119 atoms): PDB code 1hiv # inPDB file : 1hiv.PL.NativeMode.pdb (ligand: 1hiv.Lig.NativeMode.pdb) # job protocol files: MdynPar_1hiv.inp moveRes_1hiv.inp SAprotocol_1hiv.inp # Docking result potential energy file: 1hiv.ePL.DockFin.res: LigDockFin000.001.pdb:engPOTENTLG: -69.74858 !native mode LigDockFin000.002.pdb:engPOTENTLG: -56.87684 LigDockFin000.003.pdb:engPOTENTLG: -49.21509 LigDockFin001.001.pdb:engPOTENTLG: -82.47626 !best1 docking mode: ~native with disturbed end groups of Lig LigDockFin001.002.pdb:engPOTENTLG: -75.56277 LigDockFin001.003.pdb:engPOTENTLG: -58.60314 LigDockFin002.001.pdb:engPOTENTLG: -68.50492 !best2 docking mode: ~native with disturbed end groups of Lig LigDockFin002.002.pdb:engPOTENTLG: -46.25002 LigDockFin002.003.pdb:engPOTENTLG: -46.19144 LigDockFin003.001.pdb:engPOTENTLG: -18.98590 .... LigDockFin011.003.pdb:engPOTENTLG: -9.30396 LigDockFin012.001.pdb:engPOTENTLG: -59.47753 !best3 docking mode LigDockFin012.002.pdb:engPOTENTLG: -59.12785 LigDockFin012.003.pdb:engPOTENTLG: -55.48428 .... ******************************************************************************************** RESUME : 1) all shown test example of docking module successevely find a set of docking modes, i.e. files LigDockFinXXX.XXX.pdb 2) the mode with minimal potential energy of Protein-Lig interactions in the set of files LigDockFinXXX.XXX.pdb are the mode closeto the rective native ligand structure in the complex. The RMSD of the best docking mode from the native are withng 1 - 2 A. 3) The current docking method does not guarantee a finding of the best docking solution in the one RUN. The best docking solution can be obtained by refinement of the best (in the first run) docking solutions. *********************************************************************************************
CPU time = 9-10 min/1000 MD step [athlon 1400 MHz]
for protein ~ 3000 atoms
Main Program file : MDynSBmain.f
Start from the call of the input parameters
1. call inputMDSApar
reads the main Input file
filenam = './MdynPar.inp' ! in current job_dir
the file has the fixed name and located in the current job directory
the main input file MdynPar.inp defines main parameters of the job (see chapter input file description)
2. call initMolecTopSeq01
reads a defined molecular PDB file, which can be defined in the MdynPar.inp file
or has the standard name ./molec.pdb and located in the current job directory ./ ;
defines residue sequence
3. call initMolecTopSeq02
calculates 12neighbour list (covalent bonds connecting atoms) using a predefined topology
information about resdues stored in the $MDSBHOME/dat
the pair12 list array: pair12List(*) is the basic molecular topology information.
Based on the pair12List(*) the all other lists are calculated, namely
Bonded triplets and quartets to form list of covalent angles, torsion angles, improper torsion angles.
The list of triplets and quartets are calculated via tree algorithm
Call vbondListPDB2(atomXYZ,
& natom,atomNumb,atomName,resName,chName,resNumb,
& nres,resNameRes,chNameRes,
& atomNameEx,startAtInRes,
& nmoveatom,moveAtomList,
& pair12List,startPairL12,nPairL12,np12MAX,
& pair13List,startPairL13,nPairL13,np13MAX,
& pair14List,startPairL14,nPairL14,np14MAX,
& bond12List,nbond12,
& trip123List,nTrip123,np123MAX,
& quar1234List,nQuar1234,np1234MAX,
& quarImp1234L,nImp1234,nImp1234MAX)
the call of the subroutine initMolecTopPDB results in the complete definition of the molecular topology
from the input molec.pdb 3D structure.
4. call initFFieldParam
Initialization of the force field parameters for the bond, angle, torsion angle, improper angle deformations,
van der waals non bond interactions and atomic point charges for the electrostatic interactions.
For bond, angle, torsion and improper angles a respective list of parameters are generated and stored in the arrays.
A list All force field parameters are based on the amber94 force field parameter set [Cornell et.al 1995].
Molecular mechanical energy is based on the standard equations for the force field of second generation
amber94 [Cornell et.al 1995].
Decoding of the atom names (residue names) to the forceField atom name is based on the look up table
ffAtomTypeFile = $MDSBHOME/dat/atmAAmberff.dat
5. Extraction of the data from Library file
All search of the proper names in the look up table of the MDynSB program are based on
the hashing of a records in the look up table, i.e. conversion of the table in numerically
sequential order. If several records of the look up table have the same hash number (degenerated case),
they are placed in a linkedLis for this hash number.
Force field parameters are taken from the file:
ffParFile = $MDSBHOME/dat/bsparBATV.dat
code fragment to initialize force field parameters
c get ff-atom code from atomNames
call defFFatomName (ffAtomTypeFile,
& natom,atomNameEx,ResName,chName,
& ffAtomName,atomQ)
c
c define bondDef parameters for pair12List()
c
call getBondDefPar(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& bond12List,nbond12,bond12ParL)
c c define valence angles def parameters
call getVangDefPar(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& trip123List,nTrip123,ang123ParL)
c define Improper angle def parameters
call getImpDefPar(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& quarImp1234L,nImp1234,impAng1234ParL)
c define torsion parameters
call getTorsPar(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& quar1234List,nQuar1234,quar1234ParL,quar1234nPar)
c
c assign atomMass and vdwParameters
call getVDWatMass(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& nVDWtype,atomVDWtype,atomVDW12ab,atomMass)
c
c all FField Parameters are defined
6. call initSolvatGSmod
Defines atomic parameters of the current structure for the Gaussian Shell implicit solvation model [Lazaridis, 1999].
A parameters of the GS model are stored in the files:
solvGSPar_aa_amb.dat
solvGSPar.dat
7. call initMDStart(tempT0)
Initialize MD calculation:
Calculate the Initial nonBondPair lists
c generate three nonbonded atom pair Lists: van der Waals, Coulombic and solvation model.
c
makeVdW = 1
makeCL = 1
makeSL = 1
c
call initNonBondList(atomXYZ,makeVdW,makeCL,makeSL)
c
Calculates the forces on atoms for initial atomic coordinates
initial forces on atoms
c
fcall = 0
call initAllForce(fcall,atomXYZ,makeVdW,makeCL,makeSL,
& eVbondDef,vbdefForce,
& eVangDef,vAngdefForce,
& eImpDef,impDefForce,
& eTorsDef,torsAngForce,
& engVDWR1,vdwForceR1,
& engCOULR1,coulForceR1,
& engCOULR2,coulForceR2,
& restr1Eng,restr1AtForce,
& molSolEn, atomSolEn,atomSolFr)
c
Calculates initial atomic velocities, which are distributed according to Maxwell law
probability(vi) = ( ) exp(-mivi2/kT)
c
call initVelocity(temp,natom,
& nmoveatom,moveAtomList,atomMass,atomVel0)
c
8. Run MD
The subroutine mdRun perform MD run for a given number of time steps ntimeMX
c
call mdRun(ntimeMX,ntime0,ntime,ntimeR1,ntimeR2,
& ntimeF1,ntimeF2,ntimeF3,deltat,
& tempTg,tauTRF,atype,optra,wtra,nwtra,cltra)
c
9. Simulated Annealing optimization
c
call simAnnealing(nSAstep,SAProtcol)
c
with user defined SAProtocol(nstep,T) consisted of nSAstep.
Each step of the SA is MD run of nstep with particular temperature T.
All atoms of the molecular system consists of two sets of fixed
and moving atoms.
The force are calculated only for the moving atom set.
For covalent bond deformation we use the GROMOS functional form
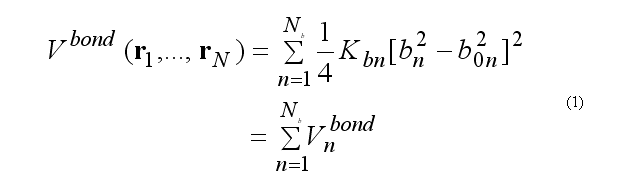
where
rij = ri - rj
bn = rij .
This functional form is equivalent to the usual harmonic function for a small deformations but a computationally is more effective.
Force on atom i due to bond n
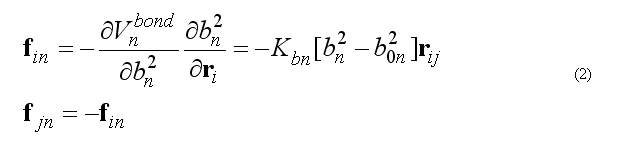
Total bond deformation force on atom i is the sum over all bonds n involving the atom i.
The calculation of the force fin is doing by
subroutine vbonddefenf(xyz1,xyz2,bondPar,edef,f1,f2) (see file vdefenforce.f)
The covalent angle deformation energy function has the form
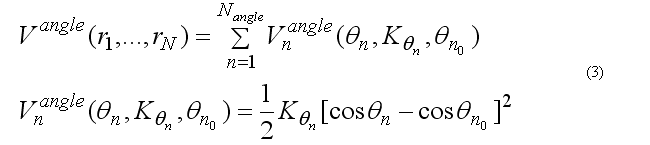
This functional form is equivalent to the usual harmonic function for the angles for a small angle deformation but a computationally is more effective. The angle 2n ( at the j ) is between atoms i-j-k . The cosine of the angle 2n

The forces on atoms i,j,k due to the deformation of the angle 2n
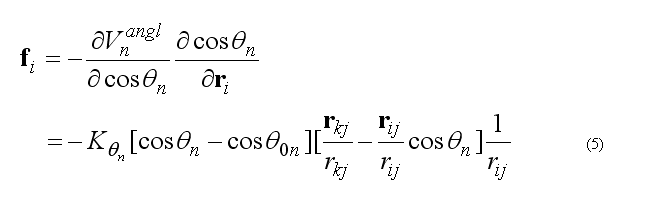
respectively force on atom k
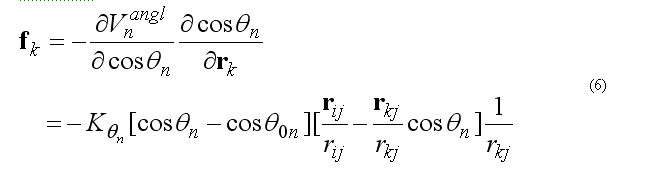
force on atom j is given from the conservation of the total force acting on three atoms

The covalent angle deformation energy and force are calculated in subroutine
subroutine vangldefenf(xyz1,xyz2,xyz3,angPar,
& edef,f1,f2,f3)
(see file vdefenforce.f)
The total torsion energy is a sum over a set of torsion angles for the four atoms i-j-k-l with a rotation around bond j-k ,
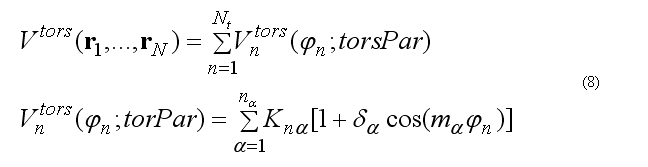
where torsion energy for bond j-k can have several torsion barriers with different multiplicity.
Torsion angle N is defined as
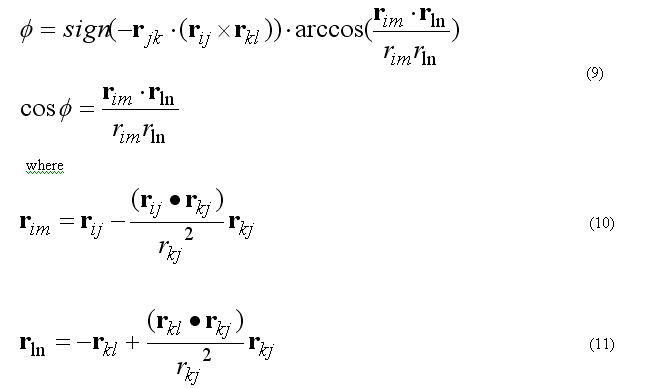
The forces on atoms i,j,k,l due to the single term of eq.(8b) are

The torsion energy and force are calculated via
subroutine torsanglenf(xyz1,xyz2,xyz3,xyz4,nTorsH,
& torsPar,eTors,f1,f2,f3,f4)
c torsPar(4*nTorsH) = {pass,Vt/2/pass,cos(delta),nFi },�
c eTors = sum{ Ki*[1+cos(delti)cos(i*Ftors)] }; i=1,..,nTorsH
c
Torsion parameters are taken from the LibData = bsparBATV.dat
The extraction of the torsion parameters from LibData = bsparBATV.dat for all quartets is done by
subroutine getTorsPar(ffParFile,
& natom,atomNameEx,ResName,chName,ffAtomName,
& quar1234L,nQuar1234,quar1234Par,quar1234nPar)
c
c InPut:
c ffParFile - ffParameters file
c natom,atomNameEx,ResName,chName : PDB info
c ffAtomName(ia) - FFatomName to search table
c the quar1234L(i),i=1,..,nQuar1234 : the QuartetList
c RESULT: quar1234Par(16*nQuar1234) - torsionFF parameters for list
c of quartets
c pass,Vt/2,delta,nFi - (printed) for each torsHarmonics,
c pass,Vt/2/pass,cos(delta),nFi - finally in array
c 4- torsionHarmanics is possible.
c quar1234nPar(iQuart) - number of torsHarmonics for the torsAngl
c
The improper torsion angle deformation keeps the four atoms 1-2-3-4 (i-j-k-l ) in specified geometry. The first atom in the improper quartet is a planar or (tetrahedral) atom. For example atoms Ci-CAi-N(i+1)-Oi are kept planar. The out of plane potential
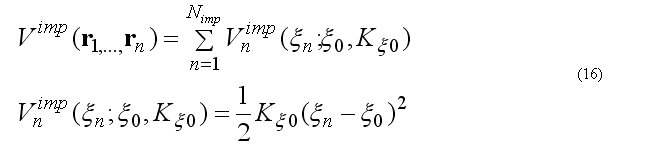
CA-N-C-CB are kept in the tetrahedral configuration (L-amino acid) or CA-C-N-CB (D-amino acid) if CA in the united atom (CH) presentation.
The out of plane angle is defined for j-i-k four atoms with i is the planar (tetrahedral)
L
angle between to planes (i-j-k) and (j-k-l) with rotation angle around j-k, other words the
torsion angle in the sequence i-j-k-l
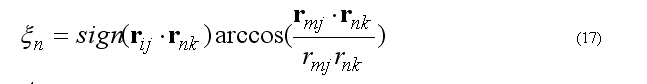
where

The forces on atoms i,j,kl due to a single term Vn
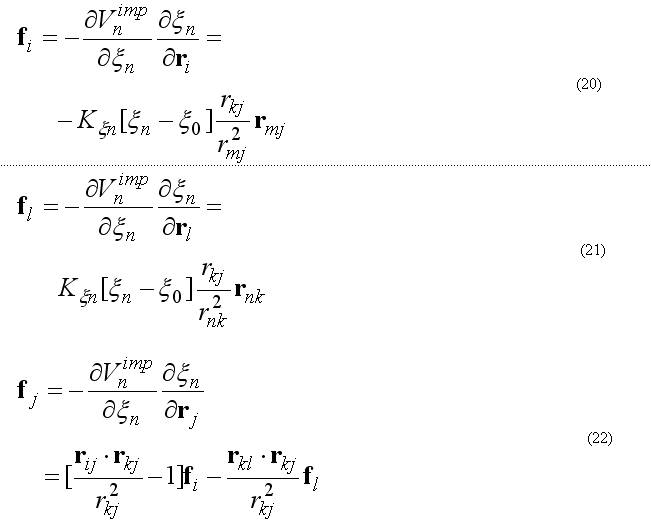
finally from the third Newton law
The improper energy and forces for a given improper quartet of atoms are calculated by the subroutine
c improper torsion energy force
c
subroutine imprtorsanglenf(xyz1,xyz2,xyz3,xyz4,impPar,
& eImpt,f1,f2,f3,f4)
c
c ImptPar(2) = K1, ksi0
All valence back-bond deformation are calculated in the file initAllForce.f
subroutine initAllForce(fcall,atomXYZ,
& makeVdWs,makeCLs,makeSLs,
& eVbondDef,vbdefForce,
& eVangDef,vAngdefForce,
& eImpDef,impDefForce,
& eTorsDef,torsAngForce,
& engVDWR1,vdwForceR1,
& engCOULR1,coulForceR1,
& engCOULR2,coulForceR2,
& restr1Eng,restr1AtForce,
& molSolEn, atomSolEn, atomSolFr)
c
include 'xyzPDBsize.h'
include 'xyzPDBinfo.h'
include 'pair1234array.h'
include 'nbondPairVCS.h'
include 'vdw12Par.h'
include 'restrainInfo.h'
include 'loopInfo.h'
include 'movingAtom.h'
include 'solvGSarray.h'
include 'optionPar.h'
c
. . . . . . . . . . . . . . . . . . . . .
c
c all GeoDef forces are calculated at each step
call allAtVBondEForce(atomXYZ,
& natom,bond12List,nbond12,bond12ParL,
& eVbondDef,vbdefForce )
c
c
call allAtVangEForce(atomXYZ,
& natom,trip123List,nTrip123,ang123ParL,
& eVangDef,vAngdefForce )
c
c
call allAtImpTEForce(atomXYZ,
& natom,quarImp1234L,nImp1234,impAng1234ParL,
& eImpDef,impDefForce )
c
c torsionEnForces
c
call allAtTorsEForce(atomXYZ,
& natom,quar1234List,nQuar1234,
& quar1234ParL,quar1234nPar,
& eTorsDef,torsAngForce )
c
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
The deformation forces are calculated at each time step in the MD run.
The non bonded pair interactions are calculated for the pair list. Pair list for the central atom i is a sequence of atom numbers for atom within the radius R from the central atom. Three separate pair lists are calculated. The Van der Waals pair list(i) includes atom j if
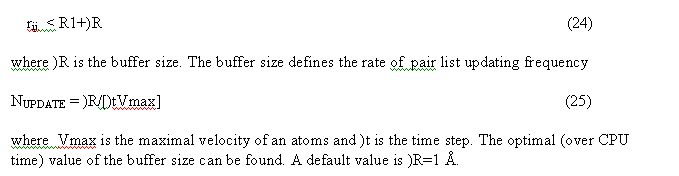
The linked list and all pairList (nnbPairLV, nnbPairLC, nnbPairLS) are calculated in the subroutine
c
subroutine nonbondListVCS(rcutV,rcutC,rcutS,atomXYZ,atomQ,
& rbuffV,rbuffC,rbuffS,
& makeVdW,makeCL,makeS,
& natom,atomNumb,atomName,resName,chName,resNumb,
& nres,resNameRes,chNameRes,
& atomNameEx,startAtInRes,
& nmoveatom,moveAtomList,moveFlag,
& pair12List,startPairL12,nPairL12,
& pair13List,startPairL13,nPairL13,
& pair14List,startPairL14,nPairL14,
& nbpairListV,startnbPairLV,nnbPairLV,nnbpLVMAX,
& nbpairListC,startnbPairLC,nnbPairLC,nnbpLCMAX,
& nbpairListS,startnbPairLS,nnbPairLS,nnbpLSMAX)
c
fragment of code for the linked list calculation:
c distribute atoms over cells
c make linked list of atoms in cells
c headat(n) - head(incellN)
c linkList(ia) - linkedList
ixm=1
iym=1
izm=1
do ia = 1,natom
c calculate cell numb
i3=3*ia-3
xyzi(1)=atomXYZ(i3+1)-xMIN(1)
xyzi(2)=atomXYZ(i3+2)-xMIN(2)
xyzi(3)=atomXYZ(i3+3)-xMIN(3)
ix = xyzi(1)/cellh+1
iy = xyzi(2)/cellh+1
iz = xyzi(3)/cellh+1
if(ixm .lt. ix)ixm = ix
if(iym .lt. iy)iym = iy
if(izm .lt. iz)izm = iz
c cell number
ncell = ix + (iy-1)*nsiz(1) + (iz-1)*nsiz(1)*nsiz(2)
if(ncell .gt. ncell3MAX)then
write(kanalp,*)'ERROR!:nonbondList: ncell3MAX is low !!'
stop
end if!
c make linked list
linkList(ia) = headat(ncell)
headat(ncell) = ia
end do !ia
c end of linked list calculation
The pair lists VDW and COULOMbic energy exclude 12, 13, 14 covalent bonded pairs. The Solvent model pairList
include all 12,13, 14 pairs.
The pair list are calculated for the range respectively:
c
rcutV2 = (rcutV + rbuffV)**2 ! range for List1 -
VDWaals - nbPairListV
rcutV2m = (rcutV - rbuffC)**2 ! range for List2 - Coulombic twin
range - nbPairListC
rcutC2p = (rcutC + rbuffC)**2 ! range for List2
rcutS2 = (rcutS + rbuffS)**2 ! range for SolvationGSList -
nbPairListS
c
see file nonbobdListVCS.f
Van der waals forces are calculated for the non-bonded pair list nbpairListV()for atoms j within rij < RCUTV the cutoff radius for van der waals interactions. The modified potential 6-12 are used
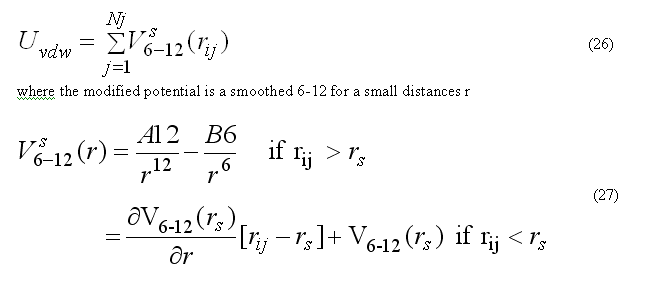
the pair list for atom i includes atoms j > i, to count each pair interaction once. The force Fvdwi on atom i due to interaction with atoms in the pair list
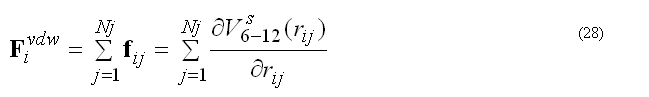
The modified (smoothed) 6-12 potential prevents over-flow when atoms are too close and generates smooth driving forces to resolve clash problems between atoms in molecular dynamics simulations, see
c subroutine vdwenforceij(dij2,dij1,rij,A12,B12,evdw,fi) c
The coulombic energy and forces for atom i are calculated for all pairs within the radius RCUTC.
The coulombic energy/forces for a central atom i are calculated for the classical coulombic law or as a coulombic interaction between two charges on the compensating background charge uniformly distributed within the sphere of radius RCUTC
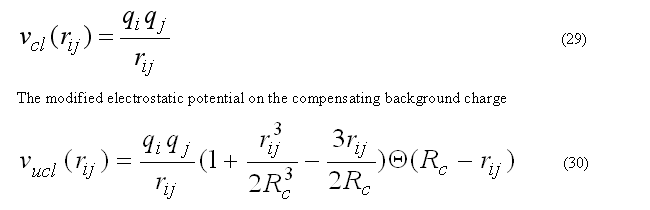
has zero interaction energy and forces for the rij > RCUTC. This form of electrostatic interactions is better suitable to prevent energy conservation in the molecular dynamic calculation, see
c
subroutine coulenforceij(var,rcutC,dij2,dij1,rij,qi,qj,ecoul,fi)
c
The nonbonded energy and force within short range RCUTV=R1 are calculated in the subroutine
c allAtNonBondEForce : VDW and COULOMBIC
c
subroutine allAtVDWEForceR1(atomXYZ,atomQ,
& natom,nmoveatom,moveAtomList,
& nbpairListV,startnbPairLV,nnbPairLV,
& pair14List,startPairL14,nPairL14,
& nVDWtype,atomVDWtype,atomVDW12ab,
& rcutV,rcutC,engVDW,vdwForce,engCOULR1,coulForceR1)
c
for the pair list nbpairListV() and pair14List(). The last one includes all 1-4 neihgbours
for which the amber force field uses the scaling factors for van der waals and coulombic interactions.
To increase performance of the van der waals energy/force calculations the table of
coefficient A12, B12 for all atom types are precalculated and then right values A12/B12 for a given
atom types in the pair ij are extracted from the vdw AB-parameter table
c get pointer to the AB table
call vdw12TablePos(nVDWtype,t1,t2,t12)
p4 = 4*t12
A12 = atomVDW12ab(p4-3)
B12 = atomVDW12ab(p4-2)
c
The long-range electrostatic forces within RCUTV < rij < RCUTC are calculated via the subroutine
c
subroutine allAtVDWEForceR2(atomXYZ,atomQ,
& natom,nmoveatom,moveAtomList,
& nbpairListC,startnbPairLC,nnbPairLC,
& rcutR1,rcutR2,engCOULR2,coulForceR2)
c
c LongRamge - RCUT1 < rij < RCUT2
The program keep separately the short-range and the long-range electrostatic energy and force.
The implicit solvation model - the Gaussian Shell model of Lazaridis & Karplus is used to calculate the solvation energy [POTEINS 35: 133-152, 1999]. The solvation free energy of the atom i
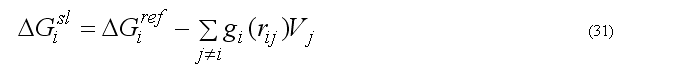
where sum is going over all neighbors of atom i which exclude volume Vj from the solvation volume around of the atom i. The function gi(r) describe the solvation energy density in the volume around the atom i and is approximated by the Gaussian function
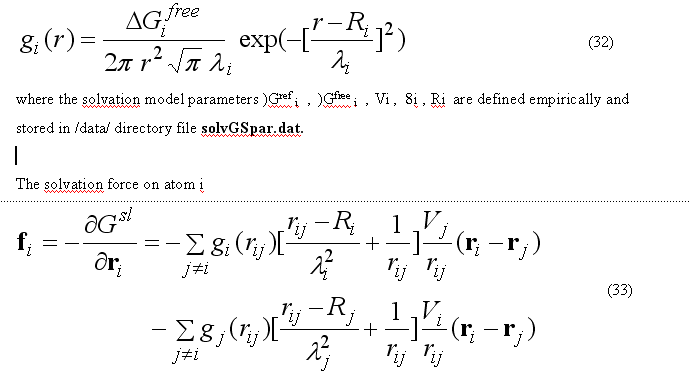
The sum over all solvation forces fi is zero.
The solvation forces are calculated by subroutine
c
call SolventEnForces(natom, atomXYZ,
& atomName,startPairL12,nPairL12,pair12List,
& nbpairListS,startnbPairLS,nnbPairLS,
& atomSolPar, molSolEn, atomSolEn, atomSolFr)
c
An MD run is performed by subroutine
c
subroutine mdRun(ntimeMX,ntime0,ntime,ntimeR1,ntimeR2,
& ntimeF1,ntimeF2,ntimeF3,deltat,
& tempTg,tauTRF,atype,optra,wtra,nwtra,cltra)
c
c MD RUN propagates MDtraj from files in mdAtomXYZvel.h
c [ atomXYZ0(*),atomVel0(*) ]
c call initMDStart(T) inits the MD start
c from the INput atomXYZ(*)-->atom0XYZ(*)
c
c ntimeMX max number of time steps
c ntime0 - executed number of timesteps in the previous call
c ntime executed number of timesteps in this call
c ntimeR1, ntimeR2 - update frequency for R1, R2 pairLists
c ntimeF1,ntimeF2 - update freq for R1=(vdw+coulR1), R2-coulR2 en/forces
c ntimeF3 - SOLVation forces
c GeoEn/force ntimeFg=1 - standart
c deltat- timestep, temp - initial(temp) of MD run
c tempTg - target T for NTV ansemble[K]
c tauTRF - tau Relaxation Factor [ps]
c atype - ansamble type = 0/1 - NEV, NTV
The MD algorithm consist of a long loop over the time steps.
For each time step MD trajectory is propagated for the )t = 1-2 femto sec,
as defined by user.
The pair lists are updated for each n-th timestep equal to ntimeR1, ntimeR2 for the short-range and for the twin-range long-range electrostatic energy calculations.
c
call initNonBondList(atomXYZ0,makeVdW,makeCL,makeSL)
c
The atomic forces due to deformation of covalent structure and short-range non-bonded
calculation are updated for the each ntimeF1-th time step, the long-range
electrostatic are updated for the each ntimeF2-th step and solvation forces are updated for each ntimeF3-th time step.
{Note! In the current version the multiple time
step for pair list update and md equation integration are equal. The general case is not tested !}
c update forces/energy
call initAllForce(fcall,atomXYZ0,doVdWef,doCLef,doSLef,
& eVbondDef,vbdefForce,
& eVangDef,vAngdefForce,
& eImpDef,impDefForce,
& eTorsDef,torsAngForce,
& engVDWR1,vdwForceR1,
& engCOULR1,coulForceR1,
& engCOULR2,coulForceR2,
& restr1Eng,restr1AtForce,
& molSolEn, atomSolEn, atomSolFr)
MD simulation can be done with a specified set of forces. The set of forces can be specified by the array fEngWF(*)
c
eGeoDef = fEngWF(1)*eVbondDef + fEngWF(2)*eVangDef
& + fEngWF(3)*eImpDef + fEngWF(4)*eTorsDef
& + fEngWF(8)* restr1Eng
engCOUL = fEngWF(6)*engCOULR1 + fEngWF(7)*engCOULR2
engPOTENT = eGeoDef + fEngWF(5)*engVDWR1 + engCOUL +
& molSolEn*fEngWF(9)
c
For one time step propagation of the MD trajectory is done by the subroutine
c make mdStep call mdTimeStepProp(nmoveatom,moveAtomList,deltat) c
which uses multi step leap-frog algorithm to calculate velocities and positions at time (t+deltat).
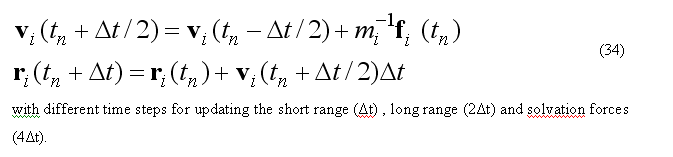
At each time step the temperature control routine performs calculation of the total kinetic energy
of the moving atoms. The relaxation the average temperature of the atomic system
to the specified value are give via the weak-coupling method or Berendsen method,
which scale the velocity by the factor lambTR(t)

Trajectory is written for each nwtra time steps. The trajectory can be written for atomic positions (and for atomic velocietis) in the user specified file.
Method description
Docking method is performed by subroutine runLigDock02 in the mdyn07 program procedure runLigDock02 perform ab initio docking of molecular ligand of size up to ~100 atoms.
The algorithm flow can be described as:
1) Calculation of the accessible surface of the protein. Calculation of a surface grid for probe sphere of radius ~ average atomic radius, and contact positions [bindSiteAt01(*)]with protein atoms. Calculation are done by subroutine surf_SAS04.
2) Calculation of a surface grid points for a probe ligand of radius of typical aromatic
ring [benzene] gridsizeSAS ~ 3.0 A. The surface grid are calculated by clustering of surface
contact positions bindSiteAt01(*) and the surface grid bindGridXYZSAS01(*) is generated. The contact
score [nsasGridPoint(*)] equal to the number of contact atomic positions included in to the surface
grid point bindGridXYZSAS01(*) is calculated.
The bindGridXYZSAS01(*) are sorted by descent of the contact score value
nsasGridPoint(*) and presents an initial trial positions for refined docking of ligand.
3) Refined docking is performed via subroutine runLigDock01(ig,bindGridXYZSAS01loc). For each initial positions bindGridXYZSAS01(*) for ligand center.
Procedure runLigDock01 perform global optimization of ligand orientation and position in a restrained region of 3D-space. Spatial restraints are a sphere of radius equal to gridsizeSAS. Orientational optimization based on exhaustive search via optimization from different initial orientations uniformly covering all orientational space. The orientational optimization can be done in two mode. Coarse grain mode consist of 24 orientations with 90deg between two neighbor orientations, fine mode consist of 144 orientations with 45deg angle between two neighbor orientations. For each initial ligand orientation the molecular dynamic simulated annealing coupled with van der waals potential scaling is performed for flexible ligand and fixed protein atoms. A variant of deformable potential energy surface global optimization method is used. Three best final position/orientations of ligand are collected for each initial positions bindGridXYZSAS01(*) in the files LigDockFinMMM.nnn.pdb - where MMM - grid position number, nnn - 001,002,003 - orientations.
The best docking variant for the ligand can be chosen as a file LigDockFinMMM.nnn.pdb with minimal potential energy engPOTENTLG:
Examples
1bty : benzamidine-trypsine complex
File #LigBindGridOnSAS: X Y Z contactScore ATOM 1 LBSt 1 16.536 26.130 8.764 11 ATOM 2 LBSt 2 29.319 14.972 16.378 11 ATOM 3 LBSt 3 6.595 15.454 32.366 9 ATOM 4 LBSt 4 28.049 26.396 3.572 9 ATOM 5 LBSt 5 37.370 14.662 29.278 8 ATOM 6 LBSt 6 9.605 28.662 39.481 7 ATOM 7 LBSt 7 18.280 35.574 15.402 7 ATOM 8 LBSt 8 30.648 34.679 44.060 7 ATOM 9 LBSt 9 34.040 33.767 21.484 7 ATOM 10 LBSt 10 5.056 19.922 18.987 6 ATOM 11 LBSt 11 25.308 5.865 13.437 6 ATOM 12 LBSt 12 13.241 31.812 30.019 6 ATOM 13 LBSt 13 6.174 15.317 15.623 6 ATOM 14 LBSt 14 15.230 11.995 39.322 6 ATOM 15 LBSt 15 42.858 27.966 33.933 6 ATOM 16 LBSt 16 39.046 14.805 5.421 5 ATOM 17 LBSt 17 24.676 37.002 14.221 5 ATOM 18 LBSt 18 39.100 25.116 6.122 5 ATOM 19 LBSt 19 25.156 6.498 5.813 5 ATOM 20 LBSt 20 14.736 13.757 2.279 5 ATOM 21 LBSt 21 35.933 31.703 11.547 5 ATOM 22 LBSt 22 45.035 21.844 22.099 5 ATOM 23 LBSt 23 12.210 8.874 28.161 5 ATOM 24 LBSt 24 11.197 11.080 32.573 5 ATOM 25 LBSt 25 25.549 16.554 -0.897 4 ATOM 26 LBSt 26 34.793 8.348 15.236 4 ATOM 27 LBSt 27 26.857 9.202 21.336 4 ATOM 28 LBSt 28 34.072 12.246 27.335 4 �..
Fig.1. Docking results for benzamidine on trypsine - 1bty complex. |
A - contact Score (black square) for binding grid points vs refined potential energy of ligand binding (red diamonds).
|
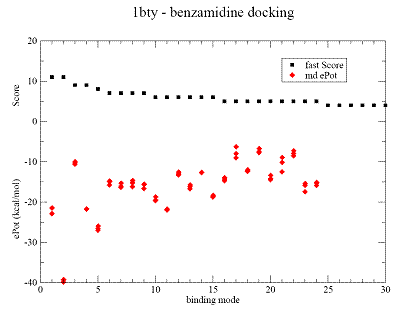 |
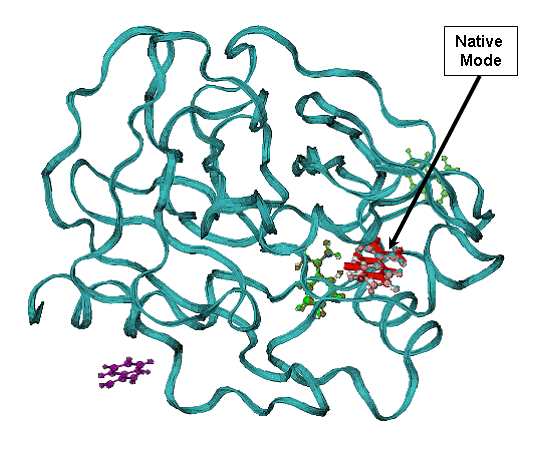
B - minimum energy docking mode (red bonds), RMSD = 0.54 A for all non Hydrogen
atoms ligand of the native binding mode.
CPK- green and violet are less favorable binding modes with low binding energy are shown in (A). CPK (pink) - native binding mode of benzamidine in 1bty. |
|
Fig.1. Docking results for benzamidine on trypsine - 1bty complex. |
A - contact Score (black square) for binding grid points vs refined potential energy of ligand binding (red diamonds).
|
|
B - minimum energy docking mode (red bonds), RMSD = 0.54 A for all non Hydrogen
atoms ligand of the native binding mode.
CPK- green and violet are less favorable binding modes with low binding energy are shown in (A). CPK (pink) - native binding mode of benzamidine in 1bty. |
|
|
Fig.2 Docking results for benzamidine on thrombin. |
A - Contact Score (black square) for binding grid points vs refined potential energy of ligand binding (red diamonds).
|
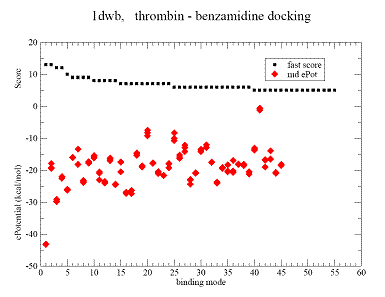 |
B(CPK blue) - minimum energy docking mode. Less favorable binding modes are shown - yellow, brown, green. CPK- (red) native benzamidine binding mode in 1dwb complex,
|
Minimum energy mode has RMSD = 0.27 A from the native binding mode of benzamidine. |
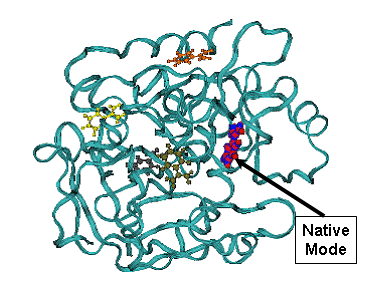
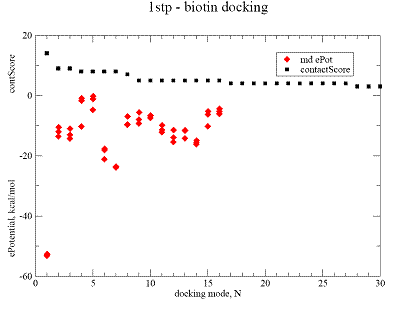
Fig.3. Docking result for biotine on streptavidine , 1stp complex. |
A - contact Score (black square) for binding grid points vs refined potential energy of ligand binding (red diamonds).
|
 |
B - minimum energy docking mode structure of biotine - CPK, lines - native biotine in the 1stp complex.
|
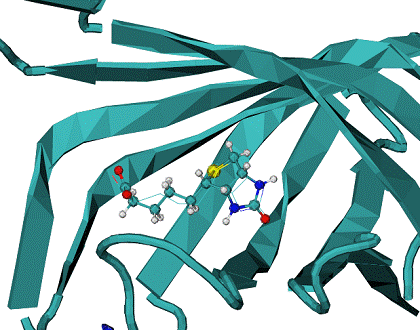
Minimum energy mode has RMSD = 0.96 A from the native binding mode of biotine. |
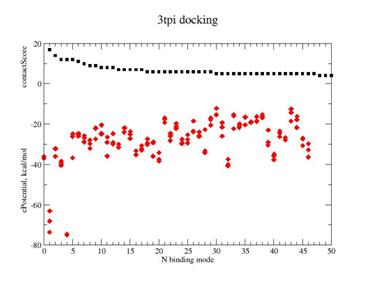
Fig. 4. Docking result for ILE-VAL dipeptide on Trypsinogen/pancreatic trypsin inhibitor. |
A - contact Score (black square) for binding grid points vs refined potential energy of ligand binding (red diamonds).
|
 |
B - Lines are minimum energy docking modes of rank 1- 4 structures of ILE-VAL peptide - lines, CPK - native binding mode of biotine in the 1stp complex.
|
The best binding energy mode has RMSD = 0.46 A from the native binding mode of dipeptide ILE-VAL |
Table 1. Energies of top ranked binding modes, and RMSD from the native binding mode.
| Binding mode | ePL, kcal/mole | RMSD, A |
| Rank 1 -
LigDockFin001.001.pdb |
-76.07 | 0.46 |
| Rank2 -
LigDockFin001.002.pdb |
-75.6 | 0.58 |
| Rank3 -
LigDockFin001.002.pdb |
75.5 | 0.78 |
| Rank4 -
LigDockFin004.001.pdb |
-74.8 | 0.88 |
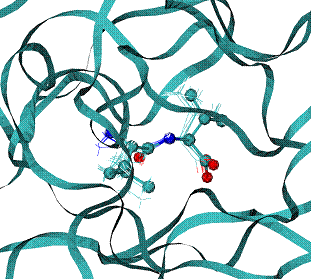
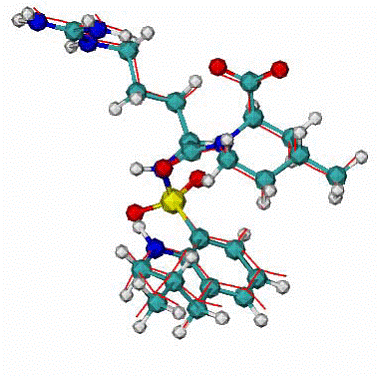
Fig. 5. 1dwc complex of Human thrombin with thrombin-inhibitor MIT . |
Human thrombin - 296 residues;
MIT - molecule includes 80 atoms |
 |
|
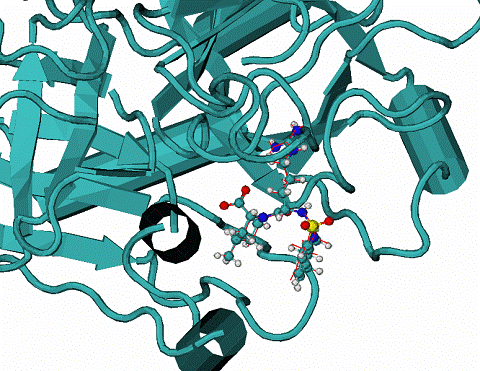
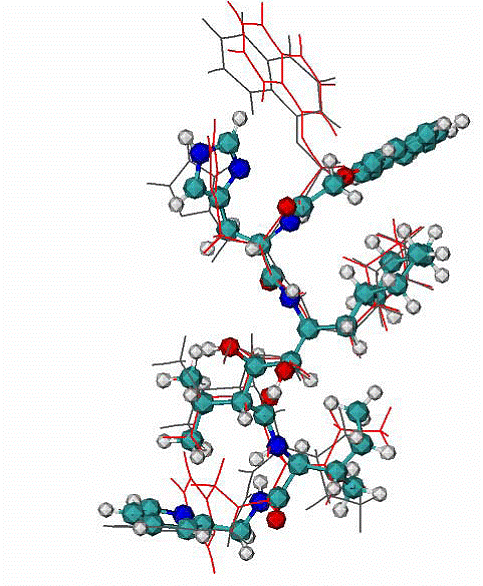
Fig. 6. 1hiv complex of HIV1 protease with inhibitor NOA
|
A -
Two top ranked calculated binding modes of NOA in comparison with the NOA ligand in the native binding mode of 1hiv complex. CPK - native binding mode, lines (red and grey) the top ranked mode by energy of binding. The RMSD from the native are ~3.1A for all atoms. The major difference between native and calculated modes are the orientation of one aromatic double-ring at the top of molecule NOA, the RMSD = 1.1. A over all atoms except the later aromatic system.
|
 |
B - 1hiv complex of HIV1 protease with inhibitor NOA CPK - native mode, red and grey lines - are calculated modes. |
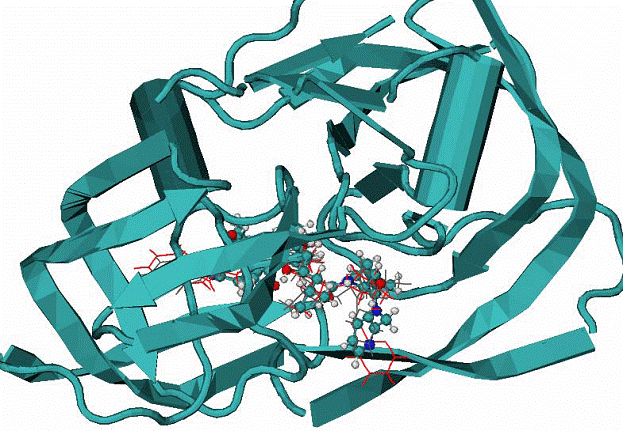
|
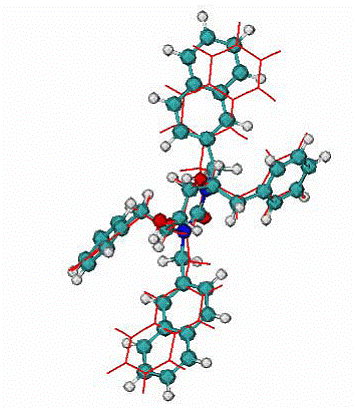
Fig. 7. 1hvr complex of HIV1 protease with inhibitor XK2
|
A -
Calculated binding mode of XK2, red lines, CPK - native binding mode of XK2 ligand. RMSD = 0.95 A for all atom.
|
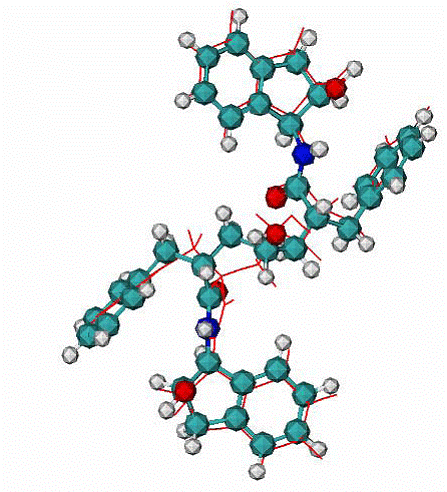
 |
B - Calculated docking mode for the ligand XK2 in complex with HIV1 protease, CPK - the native binding mode of the XK2 ligand.
|
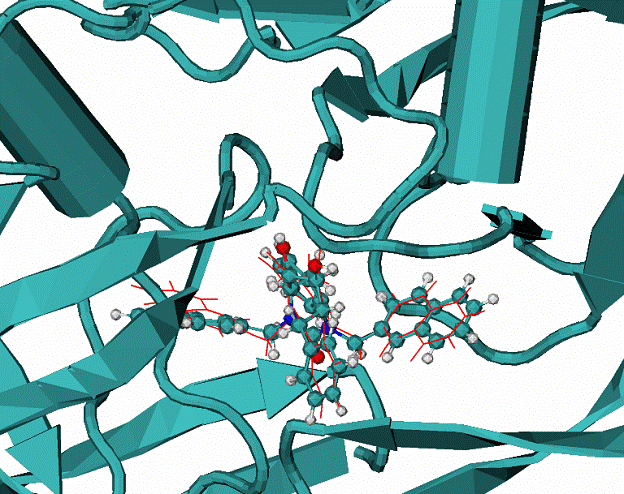
|
Fig. 8. 1hvp complex of 1HIV protease with VAC molecule inhibitor
|
A -
Calculated best binding mode of VAC is in red lines, CPK - native VAC inhibitor in the 1hvp complex; the RMSD = 0.99 A.
|
 |
B - 4hvp complex, red lines is the calculated mode, CPK - the native binding mode of VAC inhibitor.
|
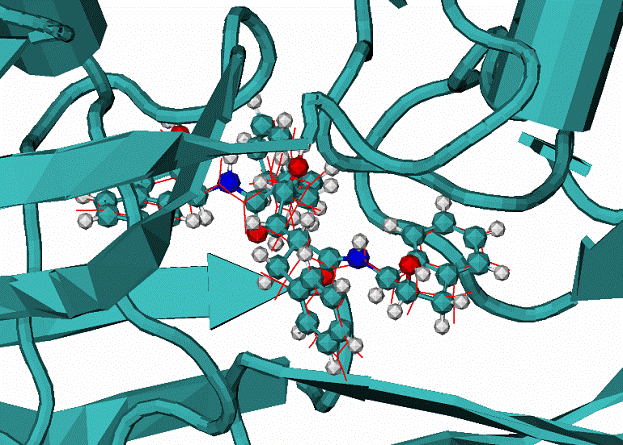
|
| Table 1. Results of MdDock method for a set of complexes | |||
| complex | Ntors | RMSD, A | DEgap |
| 1) 1bty trypsin/benz | 0 | 0.5 | 9.7 |
| 2) 1dwb a-thrombin/benz | 0 | 0.5 | 13.3 |
| 3) 1stp streptavidine/biotin | 5 | 0.96 | 29.5 |
| 4) 3tpi trypsinogen/Ile-Vla | 6 | 0.42 | 10.6 |
| 5) 1dwc a-thrombin/MIT | 8 | 0.2 | 10.8 |
| 6) 1hiv HIV1 protease/NOA | 16 | 1.1/3.1 | 2.6 |
| 7) 1hvr HIV1 protease/XK263 | 8 | 0.95 | 39.1 |
| 8) 4phv HIV1 protease/VAC | 15 | 0.9 | 3.4 |
Ntors - number of flexible torsion angles.
DEgap - energy gap between lowest energy binding mod and the next energy mode.
Conclusion:
The developed method of blind docking has show a good accuracy in prediction of the native bindig modes of flexible ligands. At the test set of 8 ligands the method shows 100% accuracy, i.e. the native binding mode are found as the mode with highest binding affinity.
Tamar Schlick. Molecular Modeling and simulation. Springer-Verlag, New York, 2000.
Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Mertz K.M.,
Ferguson D., Spellmeyer D.C., Fox T., Caldwell J.W., Kollam P.A.
A second generation force field for the simulation of proteins, nucleic acids and organic molecules.
J.Am.Chem.Soc. 1995: 117, p.5179-5197
Lazaridis T., Karplus M. Proteins: Structu, Funct., and Gen. 1999: 35, p.133-152