![]()
|
|
|
|
GetAtoms |
The program GetAtoms allow to model spatial protein structure by homology. The model of the target protein structure is built using homologous template protein structure and pairwise sequence alignment of the template and target proteins. The program allows to:
The program allows to input alignment data in various formats. The model output can be performed in PDB or AMBER formats.
For example, if we have 4hhb (A) sequence as query and 1hba(B) as template, simple format:
VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDL------SHGSAQVKGHGKKVAD
HLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPRTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLG
ALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVST
AFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVAN
VLTSKYR
ALAHKYH
Program is provided with viewer.
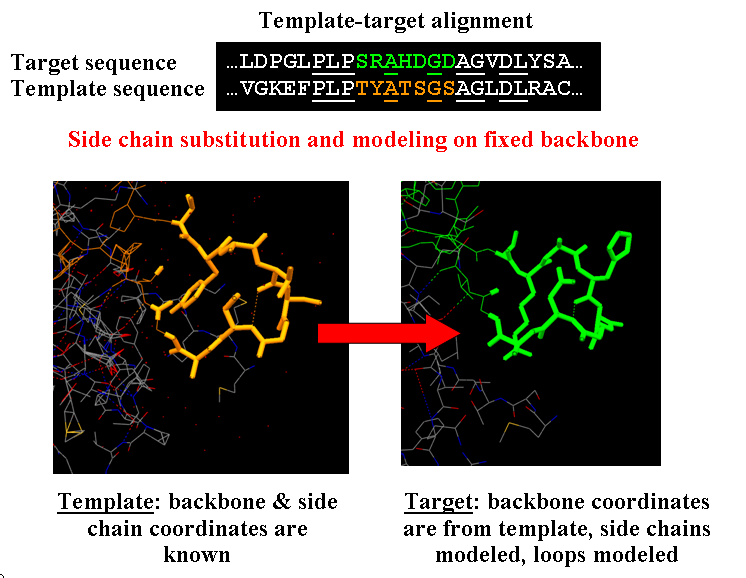
The approach is shown in the Fig.1.
The program work in three stages.
First, the program makes side chain substitution in the template structure according to amino acid sequence in the target structure. Then rough preliminary side chain optimization is performed to remove steric clashes. The optimization is performed by Monte-Carlo algorithm a nd is as follows. Initially the side chain is placed in most frequent rotameric state. Then program searches for the side chains that form clashes and try to change their conformation randomly. If the sterical energy is lower than the energy at the previous step, new configuration is accepted. If not, the energy change dE is calculated and the value of exp(-dE/Temperature) is compared to the random number rand in the range [0,1]. If rand value is lower, such conformation is accepted. The Temperature specifies the temperature for MC algorithm of side chain conformation optimization, the lower the temperature, the faster is the convergence to the nearest local minima. Higher temperature allows overcoming local minima but needing more time for search. This procedure is repeated user-defined maximal number of MC steps (for the preliminary optimization the number of 50-100 for this parameter is recommended). Sometimes the side chain rotamer configuration can be trapped in the state with high sterical energy, to overcome this, it is useful to make restart from random configuration of rotamers to new optimal configuration if optimization is not successful in 100 steps. The restart is controlled by MC process restart option.
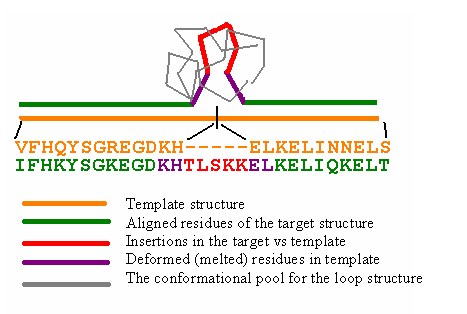
Second step performs main chain reconstruction in the insertion and deletion regions of the template-target superposition (Fig. 2).
During insertion modeling the program try to generate many loop main chain conformation in attempt to "close" the space gap between the C-terminus of the loop and N-terminus of the residue immediately following after the insertion. These conformations are generated by Monte Carlo procedure and controlled by temperature and maximal number of iteration steps as described previously. Conformations that have the distance between loop C-termini modeled N-atom and the true anchor N-atom less then user-defined threshold (C-ter attachment criterion) then screened for the conformation that have minimal sterical energy of interaction with the other part of the protein. Note, that the two template residues immediately at the place of the insertion are "melted" (actually they are added to the loop) to make local distortion in the template to allow loop to be inserted.
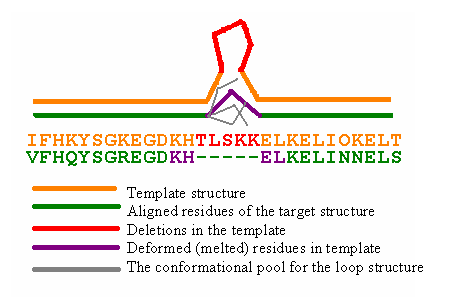
The same procedure is implemented for deletions modeling (Fig 3).
In this case two residues from both termini of the deletion are "melted" (actually they are formed a loop from 4 residues), that is build by previous algorithm.
After the insertion and deletion modeling the final optimization step is performed for side chain conformations only. The algorithm is the same as for the first step, but it is recommended to make the number of optimization steps larger (200-400).
The user can also control additional input and output parameters.
Alignment format: format of the alignment file. Several options are possible. "LOCAL", the output format of the Softberry FOLD program; "FASTA", FASTA-format; "SIMPLE", format with only sequences in the data (no sequence names); "CE", alignment format from the CE structural alignment program. First sequence is the target, second sequence is template. Columns of alignment containing only gaps in both sequences are ignored.
Adding Hydrogen atoms {ON,OFF}: the coordinates of the hydrogen atoms will be added to heavy atoms in the modeled structure.
StatusFile : the name of the file for calculation status output.
The output file contains some information about the optimization parameters and initial and final energy of the protein structure.
HEADER OXYGEN TRANSPORT 07-MAR-84 4HHB
REMARK 50
REMARK 50 GETATOMS [ver=0.9.0.0; date=20020312]
REMARK 50 Modelled from template structure provided by user.
REMARK 50 Calculation parameters:
REMARK 50 Simulated Annealing Temperature=2.000000
REMARK 50 Simulated Annealing Maximal number of steps=100
REMARK 50 Simulated Annealing steps done=-1073216864
REMARK 50 Add Hydrogen Atoms=OFF
REMARK 50 Final score data:
REMARK 50 VDW_Score=1.089206e-19
REMARK 50 Steric_Score=2.652495e-315
REMARK 50 Bump_Score=0.000000e+00
ATOM 1 N VAL 1 9.223 -20.614 1.365
ATOM 2 CA VAL 1 8.694 -20.026 -0.123
ATOM 3 C VAL 1 9.668 -21.068 -1.645
ATOM 4 O VAL 1 9.370 -22.612 -0.994
ATOM 5 CB VAL 1 8.948 -18.511 -0.251
ATOM 6 CG1 VAL 1 8.554 -18.010 -1.636
ATOM 7 CG2 VAL 1 8.176 -17.751 0.822
ATOM 8 N LEU 2 9.270 -20.650 -2.180
ATOM 9 CA LEU 2 10.245 -21.378 -3.143
ATOM 10 C LEU 2 11.419 -20.331 -4.099
ATOM 11 O LEU 2 11.252 -19.250 -5.024
ATOM 12 CB LEU 2 9.461 -22.198 -4.174
ATOM 13 CG LEU 2 8.651 -23.375 -3.627
ATOM 14 CD1 LEU 2 7.843 -24.024 -4.741
ATOM 15 CD2 LEU 2 9.576 -24.392 -2.976
ATOM 16 N SER 3 12.365 -20.722 -3.649
ATOM 17 CA SER 3 13.611 -20.183 -4.477
ATOM 18 C SER 3 14.557 -21.356 -5.125
ATOM 19 O SER 3 14.340 -22.536 -4.780
ATOM 20 CB SER 3 14.497 -19.299 -3.595
ATOM 21 OG SER 3 15.076 -20.068 -2.554
or WITH H-atoms:
REMARK 50 Add Hydrogen Atoms=ON
REMARK 50 Final score data:
REMARK 50 VDW_Score=1.089206e-19
REMARK 50 Steric_Score=2.652495e-315
REMARK 50 Bump_Score=0.000000e+00
ATOM 1 N VAL 1 9.223 -20.614 1.365
ATOM 2 CA VAL 1 8.694 -20.026 -0.123
ATOM 3 C VAL 1 9.668 -21.068 -1.645
ATOM 4 O VAL 1 9.370 -22.612 -0.994
ATOM 5 CB VAL 1 8.948 -18.511 -0.251
ATOM 6 CG1 VAL 1 8.554 -18.010 -1.636
ATOM 7 CG2 VAL 1 8.176 -17.751 0.822
ATOM 8 1H VAL 1 10.102 -20.497 1.435
ATOM 9 2H VAL 1 8.812 -20.175 2.021
ATOM 10 3H VAL 1 9.034 -21.482 1.426
ATOM 11 HA VAL 1 9.166 -20.592 -0.926
ATOM 12 HB VAL 1 10.006 -18.305 -0.091
ATOM 13 1HG1 VAL 1 9.071 -17.073 -1.845
ATOM 14 2HG1 VAL 1 8.833 -18.752 -2.384
ATOM 15 3HG1 VAL 1 7.477 -17.846 -1.671
ATOM 16 1HG2 VAL 1 7.168 -17.540 0.463
ATOM 17 2HG2 VAL 1 8.120 -18.356 1.727
ATOM 18 3HG2 VAL 1 8.686 -16.814 1.043
ATOM 19 N LEU 2 9.270 -20.650 -2.180
ATOM 20 CA LEU 2 10.245 -21.378 -3.143
ATOM 21 C LEU 2 11.419 -20.331 -4.099
ATOM 22 O LEU 2 11.252 -19.250 -5.024
ATOM 23 CB LEU 2 9.461 -22.198 -4.174
ATOM 24 CG LEU 2 8.651 -23.375 -3.627
ATOM 25 CD1 LEU 2 7.843 -24.024 -4.741
ATOM 26 CD2 LEU 2 9.576 -24.392 -2.976
ATOM 27 H LEU 2 8.525 -20.036 -1.884
ATOM 28 HA LEU 2 10.867 -22.070 -2.576
ATOM 29 1HB LEU 2 8.746 -21.553 -4.685
ATOM 30 2HB LEU 2 10.152 -22.623 -4.903
ATOM 31 HG LEU 2 7.969 -23.019 -2.854
ATOM 32 1HD1 LEU 2 7.705 -23.310 -5.553
ATOM 33 2HD1 LEU 2 8.376 -24.899 -5.114
ATOM 34 3HD1 LEU 2 6.870 -24.328 -4.356
ATOM 35 1HG2 LEU 2 9.162 -24.699 -2.016
ATOM 36 2HG2 LEU 2 9.673 -25.263 -3.625
ATOM 37 3HG2 LEU 2 10.558 -23.944 -2.822